



基因诊断发现国内首例丙酮酸羧化酶缺乏症患儿的新型剪接突变
发布日期:2022-06-15
今天跟大家分享一篇于2022年4月28日刊载在Frontiers in Pediatrics期刊(IF:3.418)上的病例报道,由成胜权教授和陶东英医生等人撰写,来自韦翰斯的杨敬敏博士参与了该项研究,先证者是一名20个月大的丙酮酸羧化酶缺乏症(PCD)患儿,系国内首个报道的PCD病例,文章探明了该患儿的新型剪接突变,并证明了其致病性;不但填补了国内PCD相关临床信息的空白,也丰富了PC基因的突变谱系,为该病的诊治贡献了新的思路。
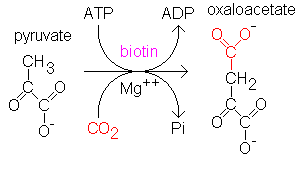
丙酮酸羧化酶(PC)作用示意图:丙酮酸羧化酶(pyruvate carboxylase)是一种线粒体基质酶,可将丙酮酸转化为草酰乙酸(oxaloacetatic acid),进而参与糖异生和能量产生的过程;
PC活性降低将导致:
①丙酮酸的积累,随后在体内转化为乳酸,最终引发乳酸性酸中毒 ;
②草酰乙酸的减少还会使糖异生发生减少,导致机体供能不足;
③其他相关的临床表型还包括酮症酸中毒和高氨血症等。
丙酮酸羧化酶缺乏症简介
丙酮酸羧化酶缺乏症(PCD)是一种罕见的常染色体隐性遗传疾病,由丙酮酸羧化酶(PC)的活性不足所致,其最为常见的临床特征包括神经发育迟缓、丙酮酸水平升高、乳酸性酸中毒、酮体水平升高和高氨血症。目前PCD主要表现为三种临床形式:
婴儿型(A型),主要在北美洲人群中发现。通常于患者出生几个月之内(婴儿期)发病,典型特征包括肌张力减退、发育迟缓和乳酸血症,随后出现感染、腹泻和其他症状,最终导致婴儿期或儿童早期死亡;
新生儿型(B型),主要在欧洲发现,尤其以法国人群为主。通常以严重的乳酸酸中毒、高氨血症为特征,患者通常于出生后3个月内死亡;
迟发型(C型)病情较轻,也称为温和型。通常表现为正常或轻度延迟的神经发育和发作性代谢性酸中毒。
截止文章成稿之时,全球范围内仅报告了33例PCD病例,而且没有一例发生在中国人群,因此本文描述的先证者为国内首例PCD患儿。
先证者情况介绍
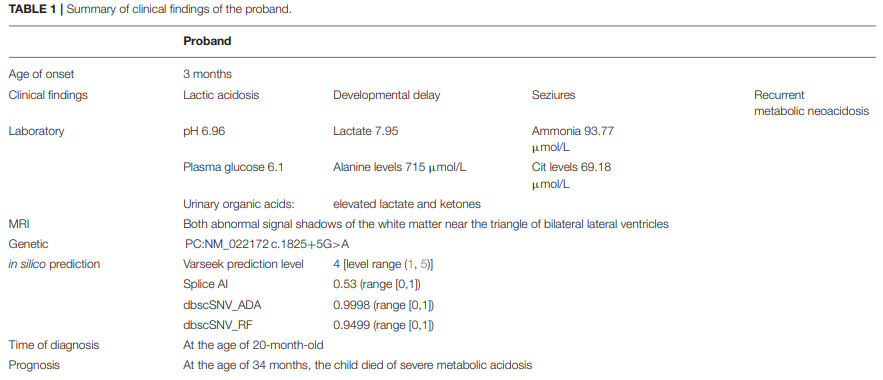
先证者是一名20个月大的男童,是一对身体健康、近亲结婚夫妻的第一个孩子,最开始因为精神发育迟缓和反复乳酸酸中毒入院。患儿足月出生,初始的身材体型均属正常,但在3个月时出现双侧眼睑阵挛发作,每次持续30秒到1分钟;成长至9个月大时,又出现了严重的乳酸性酸中毒,表现出生长迟缓、呼吸急促和嗜睡等症状,急性发作期间还会出现一系列实验室检查指标上的异常:血浆pH值降低、血浆乳酸/血浆氨和葡萄糖以及丙氨酸和瓜氨酸水平显著增高;随后在患儿34个月大时,又出现了一次极为严重的代谢性酸中毒发作,尽管进行了透析治疗,患儿的病情仍然持续恶化,乳酸水平从7.7 mmol/L上升至23.4 mmol/L,最终不治身亡。
明确致病突变
尽管先证者的生命已经无法挽回,研究者还是需要将这个宝贵的案例研究透彻,以明确国内首例PCD患儿的致病原因。通过观察患儿的临床症状已经可以初步判断为婴儿型(A型)丙酮酸羧化酶缺乏症,随后研究者通过家系全外显子组测序(WES)的手段确认了诊断;进一步的发现表明,先证者是PC基因中(c.1825+5G>A)突变的纯合子,该位点的突变在Genome Aggregation Database (gnomAD) 或1000 Genomes 数据库中均未有过报道。
随后为了验证该突变位点的致病性,研究者使用四种不同的预测软件(Varseak、splice AI、dbscSNV_ADA和dbscSNV_RF)对该突变的影响进行预测后,结果一致表明其会导致PC基因mRNA的异常剪接;
紧接着研究者对先证者及其父母,还有健康对照组的cDNA扩增产物进行了NGS测序,证实在先证者的cDNA中保留了部分PC基因13号内含子结构,从而导致蛋白编码异常,而父母双方都是相同异常剪接突变的杂合携带者;
最终使用Sanger测序与健康对照进行对比验证后,得出了可靠的结论:PC :c.1825+5G>A的突变会引起丙酮酸羧化酶基因mRNA的异常剪接,先证者样本中保留了192 bp的13号内含子结构,根据ACMG指南该变异被评定为致病;综合考虑临床表现、基因分析和cDNA测序结果,研究者最终确定该突变为患者PCD的病因。
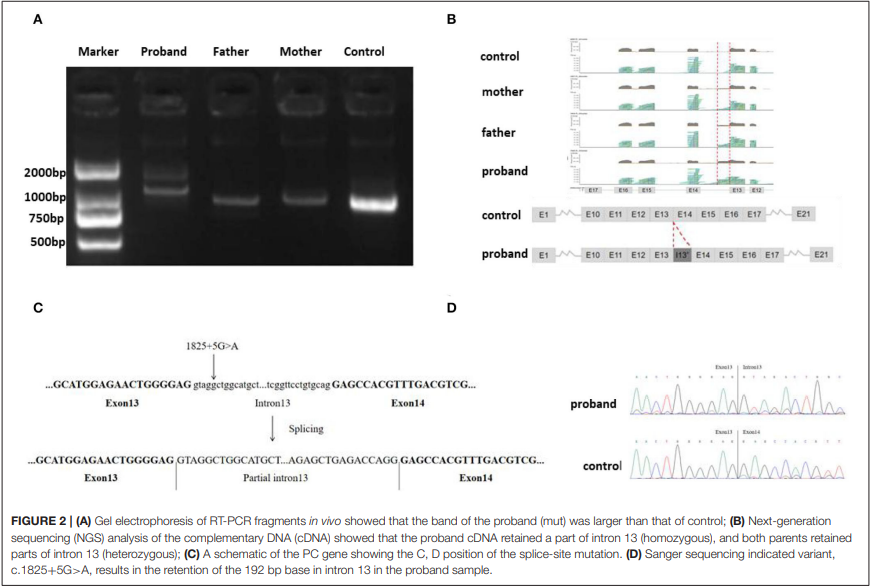
图A:体内RT-PCR片段的凝胶电泳显示,先证者的条带大于对照组,证明先证者的cDNA可能存在异常剪接;
图B:对cDNA的NGS测序结果显示,先证者的cDNA保留了部分PC基因的13号内含子且为纯合子,其父母双方均为杂合子;
图C、D:PC基因的示意图,显示出了剪接突变发生的位置以及Sanger测序结果,表明该剪切突变导致PC基因的13号内含子发生了192bp的保留。
早确诊,早治疗
对于该病的鉴别诊断,需要注意与其他遗传代谢病——尤其是容易导致高乳酸血症和神经发育异常的类别进行区分,例如I 型糖原积累综合征 (GSD1)、丙酮酸脱氢酶复合物缺乏症 (PDHCD)、遗传性果糖不耐受和果糖-1,6-双磷酸酶缺陷等等。在此过程中,一条重要的思路是发现其他疾病包括的,但是PCD表型谱中没有的一些症状,比如常见于 I 型糖原积累综合征和果糖-1,6-双磷酸酶缺乏症中的低血糖和肝肿大;或者像PDHCD与PCD在临床表型上虽然没有差异,但无法在PDHCD患者中检测出血酮体。
但是PCD本身就是极为罕见的疾病,更何况国内临床上对PCD的认知尚处于经验不足的状态,再考虑到遗传代谢病通常具有复杂的表型异质性和外显率的变化,单单通过对于患者症状的观察即使可以做到大致上的判断视其为疑似病例,但最终出于严谨和负责的角度还是有必要从基因层面进行确诊——正如文献中的研究者所做的那样。
本文报道的病例不仅是国内首例PCD病例,其突变位点还是首次发现的新型致病变异,对于该剪接突变致病性的证明不仅丰富了PC基因的突变谱系,也为此后该类疾病的基因诊断提供了新的依据。
文中提到的患儿于三个月大时表现出临床症状,初期的表现并没有呈现出很强的特异性,直到后续随着病情进展出现严重的发育迟缓和代谢异常,才引起了家属的重视送往医院治疗,但最终还是无力回天。虽然针对早期症状就做出准确的诊断困难重重,但其实并不难判断患儿的症状是由遗传病引起的,如果能够在早期就明确病因、进行治疗,多数疾病都会有更好的治疗和预后效果。
韦翰斯全外显子测序项目
(WES)
韦翰斯的全外显子测序项目(WES)可以一次检测人类基因组中近2万个已知核基因的外显子和毗邻剪接区域(±20bp),以及线粒体基因组全长测序,可同时检测线粒体突变导致的相关疾病,是针对病因不明的遗传病患者最全面、高效的解决方案。无论是发病率极低的罕见病,还是表型缺乏特异性难以诊断的遗传病,甚至像文中患儿这般是由于尚未收录的突变致病的情况,都可以通过WES的检测手段发现可能的致病突变,再经由韦翰斯认真负责的生信分析及人工解读去出具一份详实可靠的报告,把真相和希望带给患者和他们的家属。
优质服务,助力科研
韦翰斯面向各类教育研究和医疗机构提供遗传病方向的科研合作,包括基因组学、转录组学、表观遗传学和分子生物学四大方向。在了解到客户的实际需求后,我们经验丰富的管理人员会对项目的需求进行采集和评估,着手制定方案或给出方案建议,还会根据客户需要进行技术咨询;完成协商、签署合同之后,韦翰斯强大的实验团队会按照合同约定保质保量地完成项目,并且发送包括质检报告、中期报告和结题报告在内的相关结果,最终再根据客户反馈来妥善安排售后服务。
